

Blog | 5/20/2024
Implications of the FDA’s Project Optimus Initiative on the Targeted Oncology Therapy Market
Arpit Dave, PhD, Ashley Peake, Earl Gillespie, PhD, Ned Wydysh, PhD, Gary Gustavsen, Vivek Mittal, PhD
Introduction
Project Optimus is a recent, but long-coming, FDA initiative announced in 2021 to improve dosing for targeted therapies in oncology. In discussing Project Optimus, FDA notes that targeted therapies and immunotherapies have traditionally been tested in late phase trials at their maximum tolerated dose (MTD); however, increasing doses for these therapies may lead to an increase in toxicity without a corresponding increase in efficacy [1]. Dose-related post-marketing commitments are common in oncology, affecting 21% of new molecular entities (NMEs) from 2010 to 2022 [2], so Optimus should aid drug manufacturers in identifying the optimal dose earlier in clinical development and eventually reduce the number of cancer patients experiencing drug-related toxicity.
General guidance from Project Optimus includes 1) Dose escalation study design to assess pharmacokinetics (PK), pharmacodynamics (PD), safety, and efficacy at each dose level 2) Longer periods of observation for delayed toxicities 3) Leverage non-clinical data to better ensure recommended doses are in the right range 4) Consideration of multiple doses as part of later phase trials 5) Combination therapies are likely to increase toxicity and need to be investigated separate for PK, PD, safety, and efficacy 6) While the FDA acknowledges dosing in different indications may vary, they encourage information from one indication to inform dose optimization studies for a new indication [3]. While Project Optimus is not an FDA mandate, there are various regulatory and clinical levers to encourage companies’ migration away from MTD-focused study design and asset marketing. Primarily, the FDA could incentivize drug manufacturers with prioritized fast track review for assets with Optimus-compliant trial designs. In addition, FDA-mandated post-marketing dosing studies have the potential to impact guidelines and physician practice, which need to be accounted for in pharmaceutical commercialization strategies.
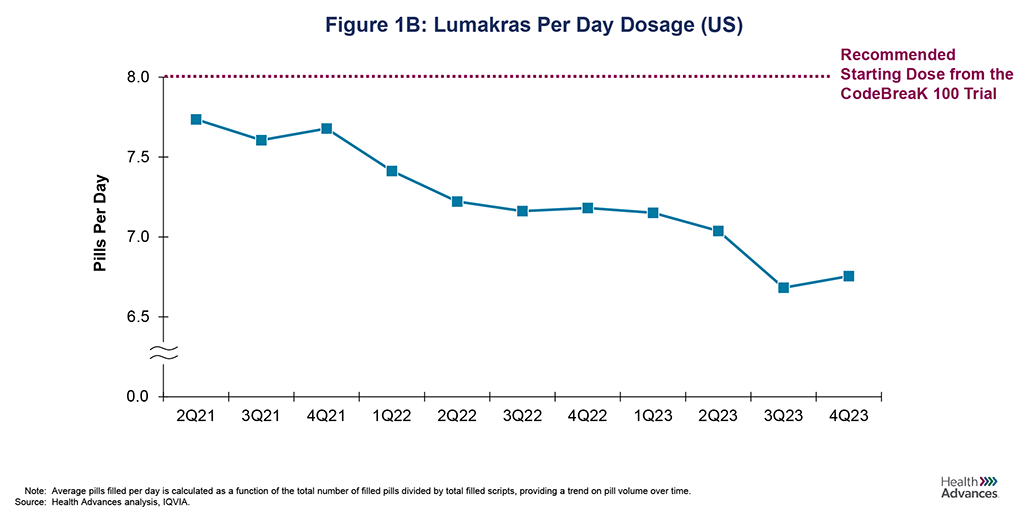
Optimus and the FDA focus on dosing are highlighted by the recent results from Lumakras’ post-marketing dosing study in KRAS G12C advanced NSCLC. The CodeBreak 100 Part B trial data showed a statistically significant decrease in toxicity when reducing the recommended dose of 960 mg/day to 240 mg/day with no statistically significant decrease in efficacy, although the 960 mg dose had a numeric advantage [4] (Figure 1A). While there has been no change to the labeled dose, analysis of claims data shows that the number of tablets an average patient is taking per day has dipped to 6.8 pills/day vs. the labeled dose of 8 pills/day (Figure 1B). While it is unclear how many commercially available drugs may be affected by post-marketing dosing studies, like Lumakras is now facing, sponsors need to consider whether there is a high risk of dose reductions driven by post-marketing dosing studies and steps they can take to mitigate that risk.
Post-Market Dose Optimization Implications on Drug Adoption
Even before Project Optimus’ implementation, there could be substantial commercial risks associated with toxicity. As targeted antineoplastics often come with substantial risk of adverse events, oncologists are familiar with managing toxicities and commonly dose reduce drugs in response to frequent or significant adverse events, or even preemptively adjust their prescribing habits for new patients. Due to their typical dose-dependent pricing structure, dose reductions can significantly impact the projected earning potential of oncology assets. In examining a few examples (Table 1), we illustrate challenges and potential solutions.
Nerlynx, a drug approved in 2017 for early-stage HER2+ breast cancer, highlights the significant impact toxicity can have on drug adoption and commercialization. Due to its severe toxicity, limited efficacy, and lower unmet need, adoption of the drug never reached initial expectations. Among a multitude of contributing factors, Nerlynx required dose-reducing patients to take fewer pills, which meant that revenue per patient declined as patients took lower doses to ameliorate toxicity. As illustrated in Figure 2, the actual revenue for Nerlynx peaked at ~$200MM, an ~85% reduction compared to the initial post-launch projections. A 2021 FDA-approved label change included the use of dose escalation to reduce toxicity, but it appears that oncologists’ minds were largely made up at that point [5]. Nerlynx stands as a clear example of a drug whose toxicity limited its commercial viability.
While Cabometyx faced similar, though not as severe, challenges to Nerlynx in dose-related toxicity, negative impacts on commercial revenue are limited in comparison [6]. Though share of prescriptions at the recommended dose of 60mg decreased from ~45% to ~15% from 2017 to 2023 (Figure 3A), the drug largely met expectations when comparing 2018 projections to actual revenues (Figure 3B). Certainly, a major difference between Cabometyx and Nerlynx is the difference in unmet need in their respective indications: while Nerlynx was in a competitive market with modest efficacy, Cabometyx was approved in areas with little competition and higher unmet need. In addition, Cabometyx was protected from revenue losses through manufacturing distinct dosages of 20mg, 40mg, and 60mg pills that are priced the same. Unlike Nerlynx, Cabometyx revenue per patient did not change regardless of the dose the patient required. This combination of unmet need, efficacy, and Cabometyx’s pricing structure mitigated commercial impacts from toxicity.
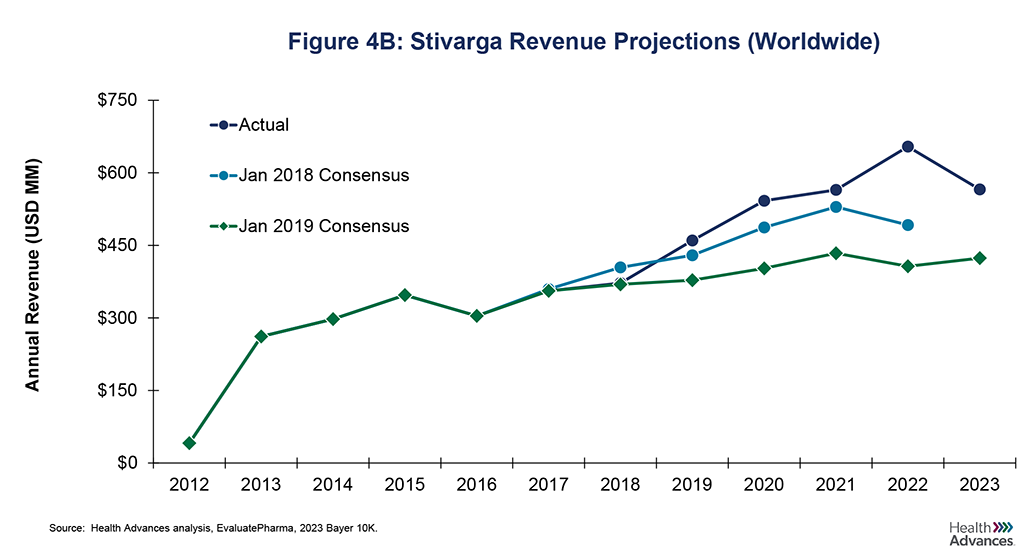
While dose reduction studies often carry commercial risks for the sponsor, there are instances where assets have surpassed forecasts despite dose reduction studies. One example is Stivarga, which was granted FDA approval in 2012 for colorectal cancer with a starting dose of 4x40mg tablets per day. Like Cabometyx, it addressed areas of high unmet need although its efficacy was more limited. After launch, revenue quickly leveled off following frequent tolerability issues (Figure 4A). In response, the ReDOS study was initiated in in 2015 [7], with study results prompting a change in NCCN guidelines in 2018. The new dosing scheme recommended a gradual increase from 80mg to 120mg, eventually reaching the maximum dose of 160mg over a period of three weeks with sufficient tolerability [8]. The more tolerable dosing strategy led to increases in patients on drug, with Stivarga unit sales in the US increasing by 14% between 2017 and 2019 and 2022 revenue that was ~60% higher than initially projected after the dose optimization study (2019 Consensus), as shown in Figure 4B. This underscores both the need for Project Optimus and the potential value of optimized dosing that could lead to increased adoption and sales.
Implications of Dose Optimization Requirements on the Targeted Oncology Pipeline
In addition to commercial risks related to dose reductions, Project Optimus’s recommendations will lead to more complex early-phase trials to gather optimized dosing data, which will increase costs and extend development timelines. While this can impact the entire breadth of the targeted oncology pipeline, smaller pharmaceutical companies will be more impacted due to their limited funding and regulatory expertise [9]. This is particularly concerning as ~70% of Phase I anti-cancer targeted therapies in the US are sponsored by smaller pharmaceutical companies (<$10B in value), as shown in Figure 5. The more substantial costs may result in increased frequency of co-sponsorship earlier in the clinical pipeline as small players look for assistance in funding larger early-phase trials. Moreover, there is concern that this added pressure on drug manufacturers could lead to the delay or deprioritization of pediatric trials [10]. This is a subgroup in which organizing trials already presents complexity and historical challenges. The FDA should consider mitigating these pressures through compensatory measures in later-stage trials or by offering additional incentives to encourage drug manufacturers to pursue the guidelines offered by Project Optimus.
Despite the evident concerns with larger investments in early-stage trials, there are some potentially compelling upsides to consider as well. With larger and more robust data from Phase I trials, the assets themselves should be more de-risked given Optimus trial measures and recommendations. Magrolimab presents an intriguing case study, highlighting the risks involved in early phase deals for assets lacking comprehensive safety data. Magrolimab is an anti-CD47 targeted therapy for hematological malignancies and was the lead asset for Forty-Seven. The drug’s promising efficacy data from an 8-month Phase 1 trial led to a lucrative $4.9 billion cash acquisition by Gilead Sciences in early 2020 [11]. This was followed by Gilead’s Phase I/II trial that concluded a combination therapy of Magrolimab with Azacitidine and Venetoclax was safe after an 8-week observation period. The trial observed a total mortality rate of 9.7% and a 19% mortality rate in the relapsed/refractory (R/R) population [12]. Gilead initiated its Phase 3 Enhance trial in 2020, with a 30.1-month observational period after randomization. However, in 2024, the trial was discontinued due to “demonstrated futility and an increased risk of death,” primarily driven by infections and respiratory failure [13]. While early-stage acquisitions are never guaranteed to be successful, they highlight the importance of robust early-phase data that could contribute to better decision-making. The FDA’s Project Optimus provides guidance on this, advocating for longer periods of observation in Phase 1 trials. By adhering to such guidance, risks can be identified earlier, potentially avoiding costly and unsuccessful acquisitions. Hopefully, this will allow for a rise in asset valuations that offset the corresponding costs and risks that Phase I sponsors take on. We look forward to tracking the shifts in the landscape of pharmaceutical investments and deals in the coming years in response to these changes.
In response to the growing use and increased scrutiny of the accelerated approval pathway, the FDA announced updated guidelines for consideration in 2023 [14]. These guidelines have tangential impacts on drugs within the scope of Project Optimus, including 1) A decrease in total accelerated approvals due to a shift in preference from single-arm trials to randomized trials 2) Project Optimus-compliant trials may receive some benefits or preferential selection for accelerated approval due to overlap in guidance and 3) Clinically relevant data generated in earlier phase trials can support later stage clinical benefit analyses. This long-term data can meet the necessary data requirements and pave the way for an expedited “one-trial” approach. The FDA’s documentation explicitly mentions these impacts, stating, “Longer term follow-up in the same trial could fulfill a post-marketing requirement to verify clinical benefit. This ‘one-trial’ approach maintains efficiency in drug development and can provide early access to a drug using the accelerated approval pathway while ensuring that a post-marketing trial is fully accrued and well underway to verify longer-term benefit in a timely fashion.” While this increased scrutiny will require additional efforts from manufacturers, there are potential opportunities that can be leveraged to better differentiate a product and improve development timelines.
Conclusion
Project Optimus presents challenges and opportunities for the healthcare industry. While there are investigational and commercial risks associated with dose-optimization, there are clear examples of success stories despite risks from dose reduction (Cabometyx and Stivarga) and there may be opportunities to avoid risky investments. Lumakras is one of the recent drugs highlighted by Optimus and the level of impact is yet undecided. On one hand the drug pricing structure for Lumakras is similar to that for Nerlynx, making it susceptible to decreased revenue from lower dosing. In contrast, it is addressing a larger unmet need than Nerlynx with less frequent toxicity. As the industry observes this story play out, we can look to historical analogs for actionable insights that drug manufacturers should be thinking about:
- Factoring in risk from potential dose reductions should play a role in drug formulation strategy. If a particular drug is susceptible to dose reduction, a pricing structure like those adopted by Exelixis for Cabometyx can limit commercial downside.
- There are opportunities to reduce patient dropouts using Optimus guidelines. Across our analogs, we see reductions in dosing prior to dose-optimization study readouts. Clearly, physician prescribing habits are impacted by more than just trial data, and FDA label or guideline advice to reduce dosing can result revenue increases if it allows more patients to tolerate a therapy.
- While dose-related toxicity can influence drug adoption and sales, meeting clinical need is still the prevailing factor determining market success. If addressing a severe patient population with limited treatment options, the threshold for toxicity is higher.
- For novel assets, develop and strategize dose-ranging studies early on, including in the preclinical stages and pre-IND FDA meetings. This can mitigate potential risks and ensure a clear transition towards the new expected guidelines.
- For existing drugs that are susceptible to dose-optimization changes, targeted physician messaging on lowered adverse events following dose reduction can be a powerful tool to support regrowing prescription volumes.
Sources
- Jain 2019 Clinical Cancer Research
- Gendy 2024 Therapeutic Innovation & Regulatory Science
- FDA 2022 Oncologic Drugs Advisory Committee Meeting
- Hochmair 2023 Annals of Oncology
- BioSpace, “FDA Approves Dose Escalation Label Update for Puma Biotechnology’s NERLYNX”
- Krens 2022 BMC Cancer
- Bekaii-Saab 2019 The Lancet Oncology
- OncLive, “NCCN Recommends Regorafenib Dose Escalation in Metastatic CRC”
- Parexel, “Three Strategies to Meet Project Optimus Requirements”
- STAT, “A Well-Meaning FDA Policy is a Threat to Pediatric Cancer Treatment”
- Gilead, “Gilead to Acquire Forty Seven for $4.9 Billion”
- Daver 2021 Blood
- Gilead, “Gilead Statement on Discontinuation of Phase 3 ENHANCE-3 Study in AML”
- FDA 2023 Considerations to Support Accelerated Approval of Oncology
::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::
Authors
Arpit Dave, PhD, is a Senior Analyst and member of the Biopharma Practice at Health Advances.
Ashley Peake is a Senior Analyst and member of the Biopharma Practice at Health Advances.
Earl Gillespie, PhD, is a Senior Director and Leader of the Biopharma Practice at Health Advances.
Ned Wydysh, PhD is a Vice President and co-leader of Health Advances’ Oncology and Cell and Gene Therapy Practices.
Gary Gustavsen is a Partner in the Precision Medicine Practice at Health Advances.
Vivek Mittal, PhD is a Partner, Managing Director, and co-leader of Health Advances’ Oncology and Cell and Gene Therapy Practices.