
Blog | 9/18/2018
New Assessment Pathway for Invasive High-Risk Medical Devices in Germany: Balancing Market Access, Evidence and Patient Benefit
By Remy Denzler, Ph.D. (Senior Analyst, Health Advances GmbH)
| Summary |
|---|
|
Background
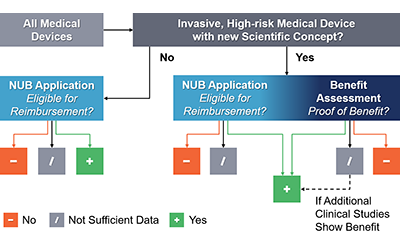
Historically, manufacturers developing novel medical devices in the inpatient segment in Germany have successfully achieved reimbursement by applying for NUB reimbursement coverage (an on-top payment above the existing DRG payment), which only necessitates a device to be novel and insufficiently reimbursed through the current system without review of clinical benefit. Driven by recent examples of new devices being ineffective or even harmful, such as a metal-on-metal hip resurfacing implant, Germany passed a new law in August 2016 that requires novel, invasive, high-risk medical devices to demonstrate a benefit to patients before being eligible for reimbursement. Benefit Assessment for Invasive, High-Risk Medical Devices The new benefit assessment applies to medical devices that fulfill the following criteria:
(1) The device is actively implanted, changes the function of, or is in direct contact with essential organ systems, and
(2) device properties result in a new area of application, or the mode of action is based on a new scientific concept.
In order to reduce market access hurdles, the new benefit assessment was set up as a parallel process to the NUB application, sharing its yearly application deadline at the end of October and its 4.5-month duration. Devices eligible for benefit assessment must present clinical evidence showing greater health outcomes for patients vs. the current standard of care in order to qualify for reimbursement.
Figure 1: Overview of the new review process for medical devices in Germany
Implications for Device Manufacturers
The new legislation has consequences for manufacturers launching a highly invasive medical device in Germany, as:
(1) there is uncertainty about device eligibility, and
(2) devices that are eligible face the risk of being excluded from insurance coverage or requiring additional clinical data.
The Federal Joint Committee (G-BA) offers a free consultation to inquire about device eligibility, and its response is legally binding and published online. As of July 2018, ~40% (5 out of 13) of examined high-risk medical devices qualified for the review process, and all devices assessed so far were either excluded from coverage or required additional data to show clinical benefit. In the latter case, the G-BA may set certain clinical study requirements. Although a device is temporarily eligible for reimbursement while collecting additional clinical evidence, the implications for manufacturers include incurrence of additional cost, loss of autonomy over trial design, and requirements to perform the investigation in German hospitals. While Germany remains an attractive market for novel medical devices, the benefit assessment legislation introduces the risks of facing more stringent clinical data requirements and of conducting a German-based clinical trial in collaboration with the G-BA.
About the Author
Remy Denzler, Ph.D. is a member of Health Advances European Practice, which helps clients navigate through the diversity of European healthcare systems to optimize commercialization strategies of pipeline and in-market products.
References (1) The Federal Joint Committee (G-BA).